Reading and understanding scientific literature can be incredibly frustrating for most people. You may want to understand some cutting-edge finding, but find you can’t wade through the technical jargon and obtuse figures, so you give up and read some crappy summary in the news. This doesn’t mean you’re not smart! I’m want to assure you that this is a learned skill–we actually have to explicitly teach our students how to do it.
I feel very strongly about making science accessible to everyone. One of the ways I’m going to do it here is to walk people through recent and exciting scientific papers. Here’s my first attempt. Please feel free to give me feedback!
Summary of Brotherton et al: Neolithic mitochondrial haplogroup H genomes and the genetic origins of Europeans. 2013. Nature Communications 4:1764.
I talked recently about how you can use genetics to test the idea that cultural changes in the past were the result of migration. A few days ago, this study was published, doing just that. I want to go through their findings, because they’re exciting and important.
Europe has a very complex prehistory, characterized by lots of migrations of different ethnic groups. Understanding this prehistory genetically is a tricky endeavor, requiring the sequencing genetic lineages of both modern and ancient populations in order to try to link them in time and space. Remember how I said that the majority of ancient DNA research targets the mitochondrial (maternal) genome? By comparing the frequency of different groups of closely related lineages (called haplogroups) in different populations, we can see how closely they are related. More distantly related populations will have different proportions of haplogroups. This is pretty intuitive when you think about the story behind the science; women living in these populations were passing down their mitochondrial lineages through their daughters and grand-daughters. When a woman moved into a new place, she would have brought her lineage with her. Populations that shared greater proportions of related women would have similar haplogroup frequencies, and would differ from more distant populations.
In modern European populations the most common haplogroup is H; it comprises something like 40% of the population. In fact, my own mitochondrial genome belongs to H, reflecting my mother’s family’s Celtic origins*. It’s therefore crucial to understand how different lineages of haplogroup H are related to each other, or what their phylogeny is. Think of a phylogeny as being analogous to a family tree, with individual mitochondrial lineages being sisters, cousins, second cousins, etc. differing by the mutations they possess. You need to work out how they’re related to each other in order to start understanding their shared histories.
Now, the phylogeny of ancient haplogroup H lineages was worked out previously, but that was done using only the hypervariable regions of the mitochondrial genome. (Again, see this post for an explanation of what the hypervariable regions are, and why they’re the targets of ancient DNA research). It turns out that there’s a whole bunch of genetic variation in the rest of the genome, and without incorporating it, the phylogeny is inaccurate.
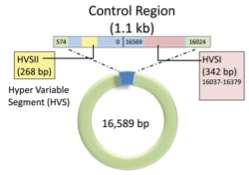
The control region, containing two hypervariable segments, makes up only a small proportion of the mitochondrial genome, but is the most frequent target of ancient DNA research. (Image modified from an original source which I’ve unfortunately lost.)
So we (finally!) get to the paper itself! What Brotherton et al. (the authors**) did was first observe that haplogroup H was much less frequent among ancient populations than in modern Europeans; Early Neolithic (~5450 BC) farmers had only a 19% frequency of H, and the older Mesolithic hunter-gatherers basically didn’t have any H. The authors decided to completely sequence the mitochondrial genomes of a sampling of ancient people who were already (through previous research) known to belong to haplogroup H. By expanding sequencing past the hypervariable regions to get at the entire genome, they would be able to “capture” all of the genetic variation, and create more high-resolution phylogenies. This would lead to a better understanding of how individual maternal lineages within H moved into the region.
They chose to sequence DNA from 37 skeletons that spanned ~ 3,500 years of the European Neolithic period (roughly 5450-1575 BC) in the Mittelelbe-Saale region of Saxony-Anhalt (Germany). Without going into the chemistry details, trust me when I say that this is a technically impressive feat!
So what did they find? I’m going to focus only on one of their main results. I’ve excerpted Figure 1A from their paper to show you:
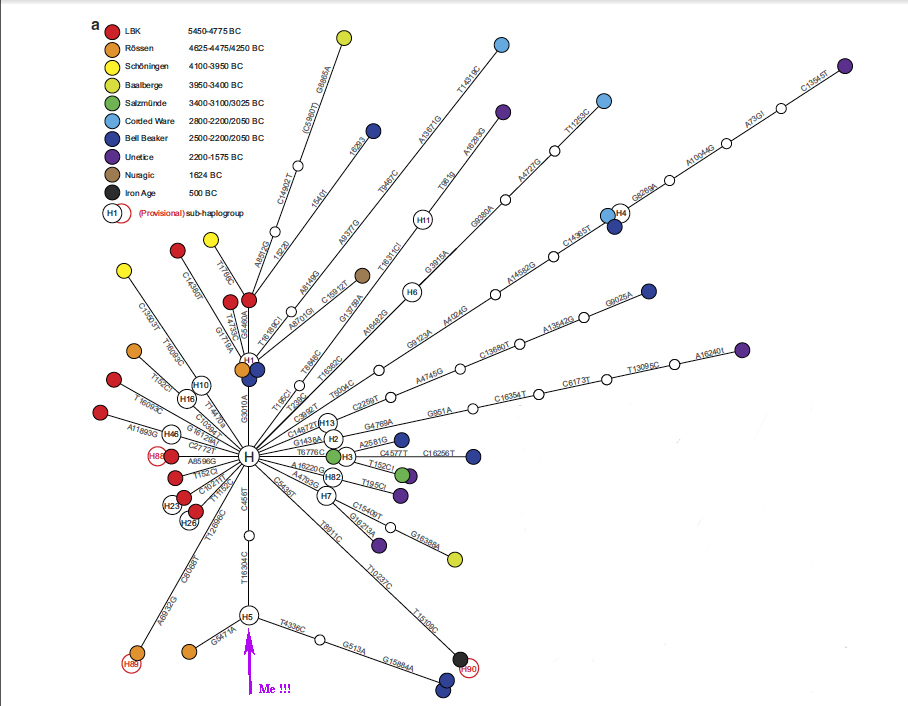
I realize this looks like something created by a demented spider. Bear with me, and I’ll explain.
This picture is a network diagram, showing the phylogenetic relationships of all the lineages they obtained from the ancient individuals. The circles are the individuals themselves, colored to represent the different cultures they come from (see the key at top left). The lines are the mutational steps between them, with longer lines indicating more mutations (and thus greater genetic distances). The mutations are listed alongside the lines. Unfilled circles are lineages which aren’t actually present at the sites, but are known about from other places. For fun, I’ve indicated with a purple arrow where I fit in on this network. (Have you ever had your mitochondrial DNA sequenced by one of the commercial genome services? If you belong to haplogroup H, see if you can find yourself on this network, too!)
How do the authors interpret this phylogeny? First, look at the position of the red circles. These are the oldest samples in the study, dating to 5450-4775 BC. Do you see how they’re on shorter lines, closer to the central node? That means they have fewer mutations away from the “basal” H type, and are therefore the oldest lineages! (Remember that lineages accumulate mutations over time, so younger, “more derived” lineages are going to have more mutations). And indeed, we see that the youngest lineages (the ones with the most mutations) tend to correspond to the more recent archaeological sites. It’s a cool pattern, that reinforces the validity of this approach.
This also shows something more subtle, but very important. We’re looking at genetic lineages present throughout time within a single region, remember? So…if that region was continuously occupied by the same group of people and their descendents, we would expect to find the oldest lineages on the same branches as the later lineages. Specifically, we’d expect to see the Early Neolithic individuals (red, orange, yellow, green) to be on the same lines (but closer to the central H node) as the Late Neolithic (light blue and blue), and the Bronze and Iron Age (brown and black) individuals. Instead, they’re all on different lines. This means they’re distinct lineages (not-very-closely-related female ancestors).
And this means that, most likely, there was considerable migration of women (and probably men, though we can’t tell from these data) into Central Europe over time, beginning around 4000 BC. The authors suggest (for various reasons which I won’t get into here) that they were likely immigrants from the West, who interacted with the early Neolithic farmers, and ultimately “superseded” their genetic diversity to shape the patterns of genetic diversity seen in present-day Europeans (including myself!). How cool is that?
Does this explanation make sense? Do you have any questions? Let me know in the comments!
————————————
*Specifically, H 5
** We have a convention for referring to a study as “So-and-so et al.” that recognizes the first author (who did most of the work). “Et al.” is short for “Et alii” which means “and the others”. It’s a cool/ pretentious bit of science tradition that reflects the discipline’s historic usage of Latin.

2 thoughts on “Science papers for non-scientists: Where do Europeans come from?”